Subproject 1: Thermodynamic properties of silicate solids and liquids and iron to the TPa range from ab-initio calculations
PI: G. Steinle-Neumann, PhD: D. Langhammer
An improved description of the internal structure of extrasolar super-Earths and its ob- servable gravitational response through the Love number k2 – which is one of the central goals of the Research Unit – requires a detailed knowledge of material properties at pressures and temperatures up to 1 TPa and 10,000 K. Here we propose to perform ab initio computations on the structure, stability and thermodynamic properties of silicate and metallic melts, as well as for silicate and oxide solids.
For the liquids, ab initio molecular dynamics simulations are performed with the goal to provide energies and pressures to fit a self-consistent thermodynamic model for the melts – formulated as a Taylor expansion of Helmholtz energy in terms of finite strain and a reduced temperature – that we previously have used successfully for liquid metals.
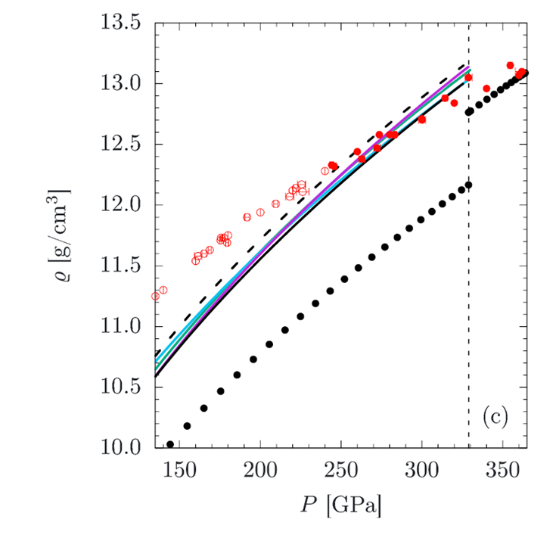
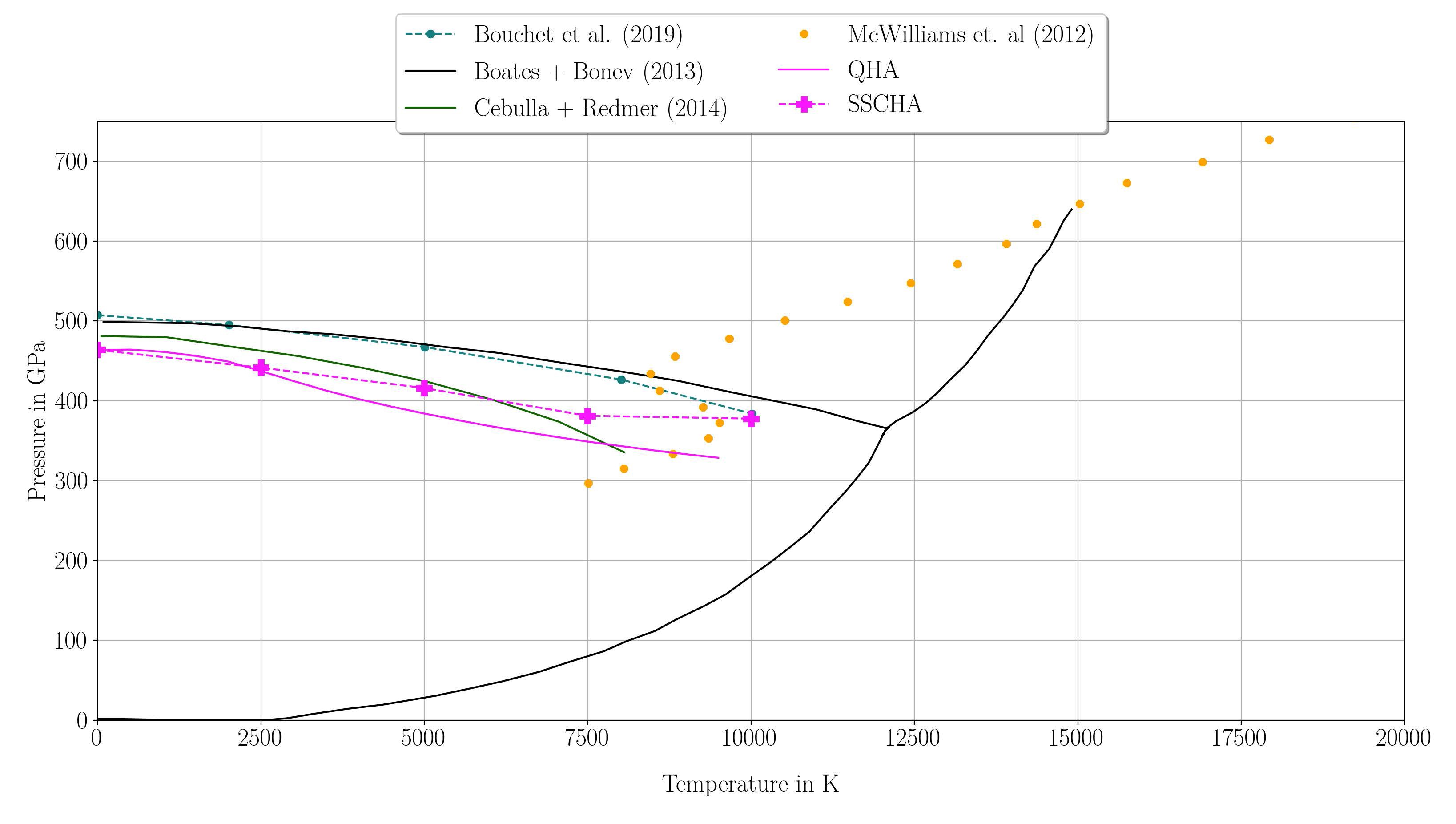
A newly established P-V-T equation-of-state of liquid iron to the TPa range (Wagle & Steinle-Neumann, 2019) shows that different models from the literature established for conditions of the Earth’s core extrapolate well to the TPa range.
Preliminary results on computing the phase transition for MgO for the B1 to the B2 phase, stable above ~500 GPa, reveal that zero-point motion shifts the phase transition to lower P, and that anharmonic contributions extend the stability range of the B1 phase to higher P.
In the second funding period, we will attempt to:
- Expand the work on phase transitions to other silicate minerals relevant for compositions of super-Earth’s.
- Establish other properties, such as the shear modulus that will affect the k2 Love number of planetary bodies.
- Formulate equations-of-state for silicate liquids potentially relevant for hot super-Earths.
- Determine viscosities for silicate liquids relevant for the magma ocean stage of planets and other melts that occur in the context of planetary evolution (e.g., volcanism).
- Consider partitioning of elements between silicate and iron liquids that determine the chemistry in different reservoirs (i.e., silicate mantles and metallic cores) of segregated planetary bodies.
Some of these properties are also relevant for the evolution and structure of the Earth, and we will also consider them in a geophysical and geochemical context to see whether we can reliably predict them.
The results obtained in SP1 will be useful in the Research Unit for guiding and interpreting experiments (SP7 and SP8) and model results can be directly used in planetary structure models (SP4).
Contact PI
Ph.D. Gerd Steinle-Neumann
Bayerisches Geoinstitut, Universität Bayreuth, 95440 Bayreuth
T: +49 (0)921 55 3702 , Email